Sierpień jest miesiącem poświęconym pogłębianiu wiedzy w społeczeństwie na temat Rdzeniowego Zaniku Mięśni, groźnej choroby, która dotyka – w zależności od typu – niemowlęta, starsze dzieci a czasem i dorosłych. Choć oficjalnie SMA należy do chorób rzadkich, jest spośród nich najczęstsza.
Bardzo ważne jest, aby zwłaszcza przyszli rodzice mieli podstawową wiedzę na temat tego schorzenia, gdyż to ich obserwacje najczęściej alarmują lekarzy. Tymczasem na świecie mamy dostępnych już kilka skutecznych terapii SMA, pod warunkiem jednak, że zostaną wdrożone bardzo szybko po wystąpieniu objawów lub nawet przed – w przypadku ustalenia wystąpienia zagrożenia chorobą poprzez wywiad lekarski i przebadania genetycznie dziecka bezobjawowego.
SMA (ang. spinal muscular atrophy), czyli rdzeniowy zanik mięśni, to ciężka choroba rzadka, w której z powodu wady genetycznej obumierają neurony w rdzeniu kręgowym odpowiadające za skurcze i rozkurcze mięśni. Brak impulsów nerwowych prowadzi do uogólnionego osłabienia i postępującego zaniku mięśni szkieletowych, a w ostateczności częściowego albo całkowitego paraliżu, także mięśni odpowiadających za oddech.


Choroba dotyka osoby w różnym wieku, jednak w ponad 90% przypadków objawy pojawiają się w niemowlęctwie albo wczesnym dzieciństwie.
SMA jest wynikiem wady genetycznej – mutacji w genie SMN1. Mutację tę osoba chora otrzymuje po obojgu rodziców, którzy zazwyczaj nie są świadomi jej posiadania. SMA nie ma wpływu na rozwój poznawczy i intelektualny. Dzieci z SMA są przeważnie bardzo inteligentne, pogodne i czerpią ogromną radość z życia.
Tradycyjnie choroba dzieli się na cztery typy lub postaci w zależności od etapu rozwoju motorycznego, na którym ją zaobserwowano, a więc pośrednio od wieku. U niemowląt i małych dzieci pierwsze objawy zwykle pojawiają się nagle, a stan zdrowia pogarsza się z tygodnia na tydzień.
W SMA1, zwanej dawniej „chorobą Werdniga-Hoffmanna”, objawy osłabienia mięśni widoczne są w pierwszych tygodniach albo miesiącach życia. Niemowlę jest wiotkie, zwykle ma osłabiony krzyk i oddech, nie jest w stanie unieść główki i nigdy nie będzie siedzieć bez podparcia. Aby przeżyć, dzieci takie wymagają wysoko wyspecjalizowanej opieki, a i tak śmiertelność w tej grupie jest wysoka.
Dzieci, które umiały utrzymać się bez podparcia w pozycji siedzącej, ale nie zaczęły chodzić samodzielnie, zaliczane są do SMA2. Również te dzieci potrzebują specjalistycznej opieki i wsparcia, chociaż ryzyko przedwczesnej śmierci jest znacznie niższe.
W przebiegu SMA3, dawniej zwanej „chorobą Kugelberga-Welander”, mówi się, jeżeli osoba była w stanie samodzielnie postawić przynajmniej kilka kroków, zanim osłabienie mięśni zmusiło ją do korzystania z wózka. Osoby z tym typem SMA zwykle prowadzą w miarę niezależne życie, choć nieraz potrzebują korzystać ze specjalistycznego sprzętu.
Gdy pierwsze objawy pojawiają się w wieku dorosłym, niektórzy lekarze nazywają to postacią czwartą SMA. Osoby z tą postacią zwykle przejawiają jedynie niedowład nóg.
Zawsze jednak SMA powoduje postępujący niedowład, osłabienie, przykurcze mięśniowe, upośledzając zdolność samodzielnego poruszania się. Niemal wszyscy chorzy na SMA poruszają się na wózkach inwalidzkich.
Dostępne terapie
W leczeniu SMA medycyna dosłownie w ostatnich kilku latach osiągnęła piorunujące postępy. Rodzice dzieci, które teraz rodzą się z wadliwym genem, mogą patrzeć w przyszłość swoich pociech z dużo większym optymizmem niż jeszcze 5-7 lat temu.
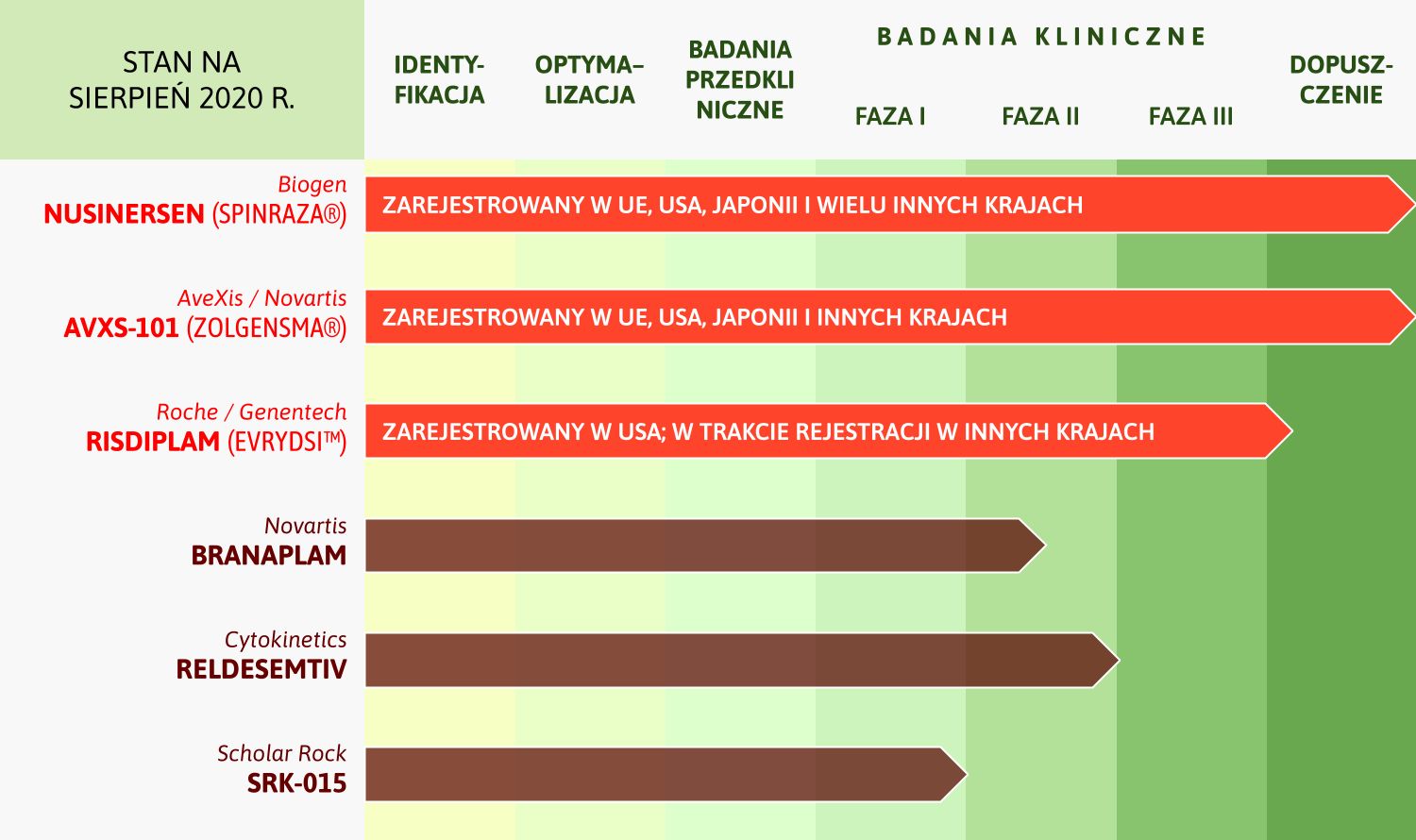
Obecnie na rynek dopuszczone są dwa nowoczesne leki przyczynowe. Ich działanie polega na zwiększeniu dostępności białka SMN w organizmie chorego:
- nusinersen (Spinraza®) – lek podawany przez wkłucie do kanału kręgowego, dopuszczony do leczenia wszystkich chorych z rdzeniowym zanikiem mięśni od grudnia 2016 r. w USA, a od maja 2017 r. w UE (a także w szeregu innych państw), refundowany w pełni w Polsce dla wszystkich chorych na SMA!
- lek terapii genowej onasemnogen abeparwowek (Zolgensma®), podawany dożylnie, dopuszczony do stosowania w USA, Japonii i na Bliskim Wschodzie do leczenia dzieci do 2. roku życia, a od maja 2020 r. dopuszczony w UE do leczenia chorych mających SMA typu 1 lub mających SMA typu 2 lub 3 w połączeniu z 2–3 kopiami genu SMN2.
Lek nie jest jeszcze dopuszczony do stosowania w Polsce, choć w kwietniu br. podano go po raz pierwszy dziecku w Polsce, dzięki warunkowemu dopuszczeniu i prywatnej zbiórce funduszy. Prawdopodobnie wkrótce i ta terapia będzie refundowana w kraju, jednak pierwszy krok w staraniach o to, należy do producenta leku, a nie ministerstwa zdrowia (jak powszechnie błędnie się zakłada, przypisując złe intencje rządzącym).
- risdiplam (Evrysdi™) – lek przyjmowany doustnie, dopuszczony w USA do leczenia chorych z rdzeniowym zanikiem mięśni i w trakcie procedury dopuszczeniowej w innych krajach
Na etapie zaawansowanych badań klinicznych znajdują się jeszcze trzy kolejne preparaty.
Obecnie skuteczność nusinersenu (Spiranzy) jest tak duża, że podanie go dzieciom przed wystąpieniem objawów choroby (na podstawie badania genetycznego stwierdzającego istnienie uszkodzonego genu) gwarantuje niemal identyczny rozwój jak zdrowych rówieśników. Oczywiście dzieci te potrzebują zabezpieczenia rehabilitacją, jednakże ich niepełnosprawność jest niewielka i będą mogły żyć samodzielnie w społeczeństwie, zakładać rodziny i być po prostu szczęśliwymi ludźmi.
Równie spektakularne efekty wydaje się – jak na razie – mieć stosowanie terapii genowej Zolgensma, na którą obecnie w Polsce prowadzi się wiele zbiórek po 8-9 mln złotych na dziecko, gdyż właśnie taka jest cena podania Zolgensmy dziecku ze SMA, dodatkowo pacjent nie może mieć więcej niż 23-24 miesiące. Ponieważ na razie refundacji tej terapii nie ma w naszym kraju, w przypadku wielu dzieci trwa wyścig z czasem (szkoda jednak, że rodzice tych maluchów często w opisach zbiórek pomijają fakt, że ich dziecko otrzymuje w Polsce za darmo nusinersen…). Zolgensma może się poszczycić jak na razie znacznie mniejszą grupą dzieci, u których zaobserwowano poprawę/stabilizację kondycji fizycznej, ale z drugiej strony poprawy stanu zdrowia bywają spektakularne także u pacjentów z zaawansowaną postacią choroby i następują szybciej niż na samym nusinersenie.
Następnym przełomowym krokiem w leczeniu SMA będzie wprowadzenie obowiązkowych badań przesiewowych noworodków w kierunku SMA, są to badania genetyczne, a więc drogie. Być może Polska będzie jednym z pierwszych krajów, które wdroży taki program, co skutkować będzie podaniem leków wszystkim dzieciom ze stwierdzoną mutacją przed wystąpieniem objawów SMA, a więc uchroni je przed głębokim kalectwem i życiem na respiratorach. Wszystkie informacje medyczne i grafika pochodzą ze strony https://www.fsma.pl/